Investigating the Difference of Gene Expression of MRSA Using Bioinformatics Tools

Investigating the Difference of Gene Expression of MRSA Using Bioinformatics Tools - Alex Feit
ABSTRACT
Background
Methicillin-resistant Staphylococcus aureus (MRSA) is a contagious staph bacterium causing skin infections that can spread to vital organs and is resistant to many antibiotics, including vancomycin. Genetic factors have been linked to susceptibility to persistent MRSA infections, providing potential insights for better treatments. This study aims to identify immunity-related genes that may contribute to resistance or susceptibility to MRSA.
Methods
Bioinformatics tools and databases were used to analyze MRSA data. Data were collected from the National Center for Biotechnology Information (NCBI) Gene Expression Omnibus (GEO), which included three groups: mock (healthy mice), mock with linezolid (a treatment), and infected with linezolid. Differentially expressed genes (DEGs) were identified by removing data with a p-value higher than 2.20e-12. The top 50 DEGs were analyzed using ShinyGO, which incorporated KEGG, Gene Ontology (GO), and SRplot tools to determine key pathways and associated genes.
Results
From 20,859 genes, the analysis identified the top 50 DEGs with significant differential expression across groups. ShinyGO revealed two key pathways: the Cytosolic DNA-Sensing Pathway and the Toll-Like Receptor Signaling Pathway. Key genes identified within these pathways included TREX1, IRF7, MLKL, CXCL10, CD14, IP-10, and MIG. These genes are associated with immune responses and regulation, offering insight into MRSA immunity mechanisms.
Conclusion
The study identified key pathways and genes that potentially contribute to MRSA immunity, consistent with previous findings. These genes can be tested in laboratory settings to explore potential antibiotics capable of targeting and weakening MRSA resistance.
KEYWORDS: Methicillin Resistant Staphylococcus Aureus infection, MRSA, NCBI, GEO, KEGG, GO, TREX1, IRF7, MLKL, CXCL10, CD14, IP-10, MIG, Cytosolic DNA-Sensing Pathway, and Toll-Like Receptor Signaling Pathway
INTRODUCTION
Methicillin Resistant Staphylococcus Aureus infection, also known as MRSA, is a very contagious type of staph bacteria that causes skin infection and is resistant to many antibiotics. The bacteria becomes dangerous when it enters the skin through a cut. Once the skin is infected, the infection can spread to the bloodstream, lungs, heart, bones, and joints (1).
MRSA is resistant to many common antibiotics and now it’s becoming resistant to its usual treatment vancomycin. Doctors are working on better ways to treat it, but MRSA keeps evolving quickly (1). Doctors are starting to think of new ways and methods to get rid of MRSA and have to keep track of it and study the differences (2)(3).
The MRSA ability to develop antibiotic resistance either happens naturally or the bacteria is able to create defense mechanisms. The antibiotics that do work, don’t work everytime. People will need multiple treatments to try to kill the disease (1). For example, MRSA is resistant to a type of antibiotic related to penicillin, cephalosporins, and carbapenems (4). Several studies have shown that genetic factors or genes make some people more prone to persistent MRSA infections, potentially paving the way for improved treatments (5).
Therefore the goal of this study is to identify genes that are related to immunity and therefore potentially identify what makes it immune. Results from this study can potentially identify genes related to MRSA’s immune system and serve as potential targets for treatment. We hypothesize that there will be differences between the expression of genes among the different mice samples; mock, mock and linezolid, infected and linezolid in during MRSA.
To conduct this study, several bioinformatics tools and databases were used. First, the National Center for Biotechnology Information (NCBI), Gene Expression Omnibus (GEO) was used to collect data on MRSA and identify the most important or top significant genes that are differentially expressed in MRSA. NCBI is a division of the National Library of Medicine (6). Gene Expression Omnibus (GEO) repository is a bioinformatics tool that archives and freely distributes gene expression experiment results conducted from the microarray method. Data in GEO represent original research deposited by the scientific community, often in compliance with grant or journal directives that require data to be made publicly available in a MIAME-supportive database (6).
The next bioinformatics tool used was the Kyoto Encyclopedia of Genes and Genomes (KEGG). KEGG is a database that helps to understand the basic principles, as well as practical utilities, of the relations between gene and genomic information and higher order functional information of genes (7). In this study, KEGG was used to determine the potential functions of the identified significant genes from the GEO bioinformatics.
Along with KEGG, the Gene Ontology (GO) bioinformatics database was used to help determine the potential functions of the identified genes. GO is the representation and processing of information about gene products and functions (8).
SRplot is a no-code bioinformatics platform that helps to run the KEGG and GO analysis without the need for computer programming (9). SRPlot is developed for scientists and biologists and supports a wide variety of graphs commonly used in biomedical and bioinformatics publications, which are integrated into a user-friendly graphic user interface (9).
Using all these different bioinformatics tools, this study on MRSA and gene expression can help provide information on what makes the bacteria so immune and identify potential immune related genes that can be used to treat the disease in the future.
METHODS
To conduct this research, the National Center for Biotechnology Information (NCBI) using Gene Expression Omnibus (GEO) bioinformatics tool was used. NCBI-GEO2R is an archive that stores and freely shares research conducted by scientists worldwide using the microarray method. (5). In the NCBI GEO search bar, “MRSA microarray.” was typed and several results were generated. One research was selected with the title, Linezolid Exerts Greater Bacterial Clearance but No Modification of Host Lung Gene Expression Profiling: a Mouse MRSA Pneumonia Model. The researchers in this study had a goal to examine gene expression differences between healthy and infected genes in mice (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE46071).

Figure 1: Methods flow chart. This flow chart shows the steps and bioinformatics tools of database Linezolid Exerts Greater Bacterial Clearance but No Modification of Host Lung Gene Expression Profiling: a Mouse MRSA Pneumonia Model.
Data Collection
Next, the provided microarray data was analyzed using the GEO2R bioinformatics tool by using a provided list of mice test subjects. The dataset I used was GSE46071. This data set was with male mice being genes being tested against healthy, healthy and with an antibiotic, and then infection and antibiotic. I divided the mice samples into three groups: mock, mock and linezolid, and infected and linezolid. Mock were the genes in the healthy mice, linezolid is a type of vaccine, and infected are the genes present in the mice infected with MRSA.
Data Analysis of GEO2R Results
To analyze this data, I used the no-code GEO2R ‘Analyze’ tool which uses pre-programmed coding using R programming language (R-Script). Multiple graphs were generated and selected graph results were analyzed. In the volcano plot, I analyzed how the genes were expressed between the different groups of samples. In the Venn diagram results, I evaluated the number of expressed genes and how many genes overlapped between the different mice sample groups.
Identification of Top 50 Differentially Expressed Genes
To identify the most important expressed genes, I used statistical analysis. I downloaded the top differentially expressed genes (DEGs) data into a Google Sheet and selected the top 50 DEGs by removing any data with a p-value higher than 2.20e-12. Top 50 DEGs).
Data Analysis using SR Plot, KEGG and GO Bioinformatics Tools and Databases
SRPlot, KEGG and GO bioinformatics tools and databases were used to find out what these top genes potentially do in the bacteria and what makes them have immunity against many antibiotics. The species in ShinyGo (ShinyGO 0.81) was set to mouse (Mouse genes GRCm39) because our data was originally collected from mice samples. The IDs of the top 50 differentially expressed genes (DEGs) were analyzed in ShinyGO bioinformatics tool and several results were generated as Gene Ontology results, and the KEGG using no-code R programming language.
RESULTS
Identification of Differentially Expressed Genes
The GEO2R bioinformatics tool was utilized to identify differentially expressed genes (DEGs). The results were visualized using a volcano plot and a Venn diagram, as shown in Figure 2. In the volcano plot comparing “infected and linezolid” versus “mock,” the genes were observed to be differentially expressed. A significant portion of the genes was highly expressed, while a notable number displayed lower expression levels. In the plot, red dots represent highly expressed (upregulated) genes, blue dots indicate less expressed (downregulated) genes, and black dots signify no significant differences between the two groups.
The Venn diagram revealed a total of 20,859 genes, with 771 genes specific to “infected and linezolid versus mock,” 552 genes specific to “mock and linezolid versus infected and linezolid,” and 3,515 genes overlapping between the two comparisons.
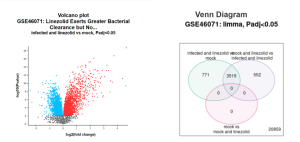
Figure 2: A and B: Data from GEO2R results A. volcano plot and B. venn diagram. In the volcano plot Red dots mean highly expressed or up-regulated. Blue dots means less express or down-regulated. And black dots mean they’re no differences between the 2 groups. Most were highly expressive, but there was still a good amount of less expressive genes. In the venn diagram, there are a total of 20859 genes. 771 of the genes are in infected and linezolid vs mock, 552 genes are in mock and linezolid vs infected and linezolid, and 3515 genes overlapped in both.
Identification of 50 Statistically Significant Differentially Expressed Genes (DEGs)
To identify my top 50 Differentially Expressed Genes (DEGs) from the total of 20859 genes, DEGs with a p-value greater than 2.20e-12 were excluded (Top 50 DEGs).
Potential Functions and Enrichment of the Identified Genes
Through ShinyGO I identified 2 pathways, Cytosolic DNA-Sensing Pathway and Toll-Like Receptor Signaling Pathway, from my KEGG Results that had with key genes of TREX1, IRF7, MLKL, CXCL10, CD14, IP-10, and MIG (Figure 3).
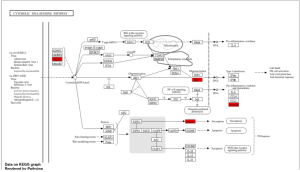
Figure 3: Key Pathway identified to be associated with the top 50 DEGs. Cytosolic DNA-Sensing Pathway with key genes highlighted in red TREX1, IRF7, MLKL, CXCL10.
Also the GO results key pathways identified were also Cytosolic DNA-Sensing Pathway and Toll-Like Receptor Signaling Pathway (Figure 4).

Figure 4: Second Key Pathway identified to be associated with the top 50 DEGs Toll-Like Receptor Signaling Pathway with key genes highlighted in red; CD14, IRF7, IP-10, and MIG
In summary, the key genes in the KEGG results were TREX1, IRF7, MLKL, CXCL10, CD14, IRF7, IP-10, and MIG and key pathways from GO results is Cytosolic DNA-sensing pathway an
DISCUSSION
Some studies have shown that genetic factors make some people more prone to persistent MRSA infections, potentially paving the way for improved treatments. For example, one study found that a mutation in DNMT3A, an enzyme involved in regulating gene expression through DNA methylation, helps fight MRSA infections. It does this by boosting gene regulation and reducing levels of the anti-inflammatory cytokine IL-10 (5).
The goal of this study was to identify genes in MRSA that are related to immunity (Figure 3 and 4) and the differentially expressed genes in the groups as mock, mock and linezolid, and infected and linezolid (Figure 2). My hypothesis was that the genes were differentially expressed among the different groups. Most of the genes were highly expressed making it react to change easily and its key genes and pathways helped it to be immune.
The findings of the key genes and pathways from this study (Summary Table 1) are supported with many other studies stating MRSA immunity (1). First, results identified TREX1 (Figure 3 ). Previous studies have shown that indeed TREX1 helps the MRSA immune system not attack its own DNA (9). Next, this study identified IRF7, which is an important transcriptional regulator of cellular responses in MRSA (10). MLKL helps the MRSA fight off infections by killing infected cells (11) CXCL10 sends signals to certain cells, telling them where to go and what to do to fight off infections or diseases (in this case vaccines and antibiotics) (12). CD14 is an important molecule that helps activate the body’s first line of defense against infections that is found either attached to the surface of immune cells or floating freely in the body (13). IRF7 is an important transcriptional regulator of cellular responses in MRSA (10). IP-10 is a special protein that gets released when the body is fighting off infections (14). Mig is a special protein that is released when the MRSA gets a signal from interferon-gamma and acts like a “signal” that attracts activated T cells (15).
In the real world, we can use this information on creating vaccines that can target MRSA immunity. Below is a summary of the key genes and pathways identified in this study that are linked to MRSA and with a potential for targets in vaccine development.
Table 1. Genes and Pathways Identified in this Study
Gene | Pathways | Connection to MRSA |
TREX1 | Cytosolic DNA-Sensing Pathway | TREX1 helps the MRSA immune system not attack its own DNA (10). |
IRF7 | Cytosolic DNA-Sensing Pathway | IRF7 is an important transcriptional regulator of cellular responses in MRSA (11). |
MLKL | Cytosolic DNA-Sensing Pathway | MLKL helps the MRSA fight off infections by killing infected cells (12) |
CXCL10 | Cytosolic DNA-Sensing Pathway | CXCL10 sends signals to certain cells, telling them where to go and what to do to fight off infections or diseases (in this case vaccines and antibiotics) (13). |
CD14 | Toll-Like Receptor Signaling Pathway | CD14 is an important molecule that helps activate the body’s first line of defense against infections that is found either attached to the surface of immune cells or floating freely in the body (14). |
IRF7 | Toll-Like Receptor Signaling Pathway | IRF7 is an important transcriptional regulator of cellular responses in MRSA (11). |
IP-10 | Toll-Like Receptor Signaling Pathway | IP-10 is a special protein that gets released when the body is fighting off infections (15). |
MIG | Toll-Like Receptor Signaling Pathway | Mig is a special protein that is released when the MRSA gets a signal from interferon-gamma and acts like a “signal” that attracts activated T cells (16). |
Limitations
One limitation is that the identified genes will need to be further studied by scientists , since we used bioinformatics datasets from microarray experiments conducted by other researchers. These identified genes can be tested in the laboratory by scientists to find what antibiotics can weaken the gene.
References
- “MRSA Infection – Symptoms & Causes.” Mayoclinic, Mayo Foundation for Medical Education and Research, 8 Nov. 2022, www.mayoclinic.org/diseases-conditions/mrsa/symptoms-causes/syc-20375336.
- Rasmussen RV, Fowler VG Jr, Skov R, Bruun NE. Future challenges and treatment of Staphylococcus aureus bacteremia with emphasis on MRSA. Future Microbiol. 2011 Jan;6(1):43-56. doi: 10.2217/fmb.10.155. Erratum in: Future Microbiol. 2011 Feb;6(2):250. PMID: 21162635; PMCID: PMC3031962.
- Mason A, Foster D, Bradley P, Golubchik T, Doumith M, Gordon NC, Pichon B, Iqbal Z, Staves P, Crook D, Walker AS, Kearns A, Peto T. Accuracy of Different Bioinformatics Methods in Detecting Antibiotic Resistance and Virulence Factors from Staphylococcus aureus Whole-Genome Sequences. J Clin Microbiol. 2018 Aug 27;56(9):e01815-17. doi: 10.1128/JCM.01815-17. PMID: 29925638; PMCID: PMC6113501.
- “MRSA (Methicillin-resistant Staphylococcus aureus).” Cleveland Clinic, my.clevelandclinic.org/health/diseases/11633-methicillin-resistant-staphylococcus-aureus-mrsa. Accessed 7 May 2024.
- F. Mba Medie, B.K. Sharma-Kuinkel, F. Ruffin, L.C. Chan, M. Rossetti, Y. Chang, L.P. Park, A.S. Bayer, S.G. Filler, R. Ahn, E.F. Reed, D. Gjertson, M.R. Yeaman, V.G. Fowler, , Genetic variation of DNA methyltransferase-3A contributes to protection against persistent MRSA bacteremia in patients, Proc. Natl. Acad. Sci. U.S.A. 116 (40) 20087-20096,https://doi.org/10.1073/pnas.1909849116 (2019).
- Tanya Barrett, Stephen E. Wilhite, Pierre Ledoux, Carlos Evangelista, Irene F. Kim, Maxim Tomashevsky, Kimberly A. Marshall, Katherine H. Phillippy, Patti M. Sherman, Michelle Holko, Andrey Yefanov, Hyeseung Lee, Naigong Zhang, Cynthia L. Robertson, Nadezhda Serova, Sean Davis, Alexandra Soboleva, NCBI GEO: archive for functional genomics data sets—update, Nucleic Acids Research, Volume 41, Issue D1, 1 January 2013, Pages D991–D995, https://doi.org/10.1093/nar/gks1193
- Minoru Kanehisa, Susumu Goto, Shuichi Kawashima, Akihiro Nakaya, The KEGG databases at GenomeNet, Nucleic Acids Research, Volume 30, Issue 1, 1 January 2002, Pages 42–46, https://doi.org/10.1093/nar/30.1.42
- Smith B, Williams J, Schulze-Kremer S. The ontology of the gene ontology. AMIA Annu Symp Proc. 2003;2003:609-13. PMID: 14728245; PMCID: PMC1480173.
- Turner NA, Sharma-Kuinkel BK, Maskarinec SA, Eichenberger EM, Shah PP, Carugati M, Holland TL, Fowler VG Jr. Methicillin-resistant Staphylococcus aureus: an overview of basic and clinical research. Nat Rev Microbiol. 2019 Apr;17(4):203-218. doi: 10.1038/s41579-018-0147-4. PMID: 30737488; PMCID: PMC6939889.
- Yan N. Immune Diseases Associated with TREX1 and STING Dysfunction. J Interferon Cytokine Res. 2017 May;37(5):198-206. doi: 10.1089/jir.2016.0086. PMID: 28475463; PMCID: PMC5439420.
- Qing F, Liu Z. Interferon regulatory factor 7 in inflammation, cancer and infection. Front Immunol. 2023 May 12;14:1190841. doi: 10.3389/fimmu.2023.1190841. PMID: 37251373; PMCID: PMC10213216.
- Tovey Crutchfield EC, Garnish SE, Hildebrand JM. The Role of the Key Effector of Necroptotic Cell Death, MLKL, in Mouse Models of Disease. Biomolecules. 2021 May 28;11(6):803. doi: 10.3390/biom11060803. PMID: 34071602; PMCID: PMC8227991.
- Liu M, Guo S, Stiles JK. The emerging role of CXCL10 in cancer (Review). Oncol Lett. 2011 Jul;2(4):583-589. doi: 10.3892/ol.2011.300. Epub 2011 May 9. PMID: 22848232; PMCID: PMC3406435.
- Y. Tesfaigzi, M. Daheshia, CD14, Editor(s): Geoffrey J. Laurent, Steven D. Shapiro, Encyclopedia of Respiratory Medicine, Academic Press, 2006, Pages 343-347, ISBN 9780123708793, https://doi.org/10.1016/B0-12-370879-6/00063-6.
- Sasya Madhurantakam, Zachary J Lee, Aliya Naqvi, Shalini Prasad, Importance of IP-10 as a biomarker of host immune response: Critical perspective as a target for biosensing, Current Research in Biotechnology, Volume 5, 2023, 100130, ISSN 2590-2628, https://doi.org/10.1016/j.crbiot.2023.100130.
- Sgadari C, Farber JM, Angiolillo AL, Liao F, Teruya-Feldstein J, Burd PR, Yao L, Gupta G, Kanegane C, Tosato G. Mig, the monokine induced by interferon-gamma, promotes tumor necrosis in vivo. Blood. 1997 Apr 15;89(8):2635-43. PMID: 9108380.

