Investigating the Replication of Sars Cov 2 Virus Using Bioinformatics

Investigating the Replication of Sars Cov 2 Virus Using Bioinformatics - Rohan Kuruganthy
ABSTRACT
Background
SARS-CoV-2 is the virus that causes the disease, COVID-19. As we know, COVID-19 has caused many deaths and changed the world as we know it. According to worldometers.info, Sars Cov2 killed 7,010,681 people and diagnosed 704,753,890 people. How did the virus replicate so fast?
Purpose
The purpose of this study was to identify genes that are differentially expressed and potentially linked to the rapid replication of SARS-CoV-2.
Methods
In this study, we utilized a variety of bioinformatics tools and databases to identify differentially expressed genes (DEGs) and their potential functional and biological pathways. We used NCBI – GEO2R and ShinyGO bioinformatics tools. Specifically we studied the microarray GEO Dataset GSE268196 to identify and analyze genes that are expressed differently between infected and uninfected samples using NCBI’s GEO2R bioinformatics tool. Then to predict the functions or associations of these genes, we used the ShinyGO bioinformatics tool to conduct Gene Ontology (GO) enrichment analysis. Furthermore, we performed KEGG enrichment analysis to determine the biological pathways in which these DEGs are involved and their potential contribution to the rapid replication of SARS-CoV-2.
Results
GEO2R bioinformatics analysis showed that various genes were differentially expressed between infected samples and uninfected samples, and the top 39 differentially expressed genes (DEGs) were identified using statistics. Further analysis of the top 39 differentially expressed genes showed that these genes are mostly enriched in several pathways related to inflammation, including mitogen-activated protein (MAP) kinase (MARK), ERK1/2 Mitogen-activated proteins and protein kinase during infection with SARS-CoV-2.
Conclusion
This study reveals the involvement of several proteins and pathways related to inflammation; mitogen-activated protein kinase (MAPK) during infection with SARS-CoV-2. Because COVID-19 often leads to uncontrolled inflammatory responses such as MAPK pathways, the pathways and proteins found to be enriched in our study can potentially be used in future studies to provide customized anti-inflammatory treatment to COVID-19 patients alongside the regular early use of antiviral drugs.
KEY WORDS
GEO2R, ShinyGO, NCBI, GEO, SARS-CoV-2
INTRODUCTION
The virus that causes COVID-19 called severe acute respiratory syndrome coronavirus (SARS-CoV-2) has caused many deaths and changed the world as we know it (Hu et al, 2021). SARS-CoV-2 can be caught from the air, physical contact, and through spit (Hu et al, 2021). According to worldometers.info, Sars Cov2 killed 7,010,681 people and diagnosed 704,753,890 people. How did the virus replicate so fast?
The problem this research is addressing is the question; why does SARS-CoV-2 replicate so fast and what genes are differentially expressed during infection with SARS-CoV-2 virus? In order for viruses to infect an organism and spread, they need to replicate (V’kovski, P et al, 2021). Therefore, one way to successfully find a cure or medicine to treat viral infections is by understanding why viruses replicate.
SARS-CoV-2 replicates in the lungs and results in various clinical outcomes, ranging from cases with no symptoms, to individuals nearing death (Wiersinga et al., 2020).
Specifically SARS-CoV-2 infections can develop into very serious lung injuries, inflammation and even death (V’kovski, P et al, 2021, Cusato et al, 2023). In severe cases, an excessively active immune response leads to harmful inflammation. This inflammation affects various tissues and cell types, including those not infected by the virus, and resembles the inflammation observed in certain autoimmune diseases (Zhang et al, 2024).
The tragic COVID-19 pandemic led many researchers to carry out replication studies on SARS-CoV-2. For a virus to replicate successfully and cause disease in an organism, six main steps are involved. First the virus attaches to the organism or host, then it penetrates the organisms or host. Following penetration into the organism, it removes its outer protein structure (uncoats) and then it replicates to spread inside the organism. Following replication, the virus assembles itself and then releases its genetic material or virion inside the organism to cause disease (V’Kovski Pet al 2021). According to previous studies, during infection with SARS-CoV-2, over 300 human proteins have been found to interact with SARS-CoV-2. Researchers have hypothesized that if these interactions between human and viral proteins were blocked, the replication of the virus can be stopped and this can stop the virus from being passed on from one person to another to cause the pandemic (V’kovski, P et al, 2021).
Therefore, the goal of this study is to identify genes that are expressed differently (DEGs) during infection with SARS-CoV-2 and the potential functions and biological pathways that may explain how SARS-Cov-2 replicates so fast. We hypothesized that the SARS-CoV-2 virus replicates by taking over the host’s gene expression machinery and upregulating genes related to replication.To address the hypothesis, this research used different bioinformatics tools and databases to find the genes in SARS-CoV-2 that could be targets for antiviral medicine against COVID-19. Specifically, this study used NCBI’s GEO2R bioinformatics tool and database (https://www.ncbi.nlm.nih.gov/geo/geo2r/) to find a dataset that contained SARS-Cov-2 gene expression data, and gene enrichment bioinformatics tools to determine gene functions and biological pathways associated with the identified genes.
The research is important because the genes and or biological pathways identified in this research can then be used by scientists to potentially develop additional antiviral medication or vaccines that can stop the virus from replicating and thus stop it from spreading.
METHODS
We used bioinformatics tools, NCBI’s GEO (https://www.ncbi.nlm.nih.gov/geo/geo2r/) and ShinyGO (http://bioinformatics.sdstate.edu/go/). GEO was used to identify genes that are expressed differently between infected samples and control samples. Then to analyze the different genes further, ShinyGo bioinformatics tool (http://bioinformatics.sdstate.edu/go/) was used. The methodology used in this study is summarized in Figure 1.
Identification of differentially expressed genes
To identify which genes are differentially expressed during SARS-CoV-2 infection, GEO2R within the NCBI Bioinformatics database was used. Specifically, the GEO Dataset: GSE268196 was used. In this dataset, the scientists took lung cell samples from monkeys infected with Sars Cov2 on days 0, 3, 7, 14, and around a month after infection. They have 71 samples in total. These samples are divided into three groups: ALFNg (22 samples), ALL10 (24 samples), and control (24 samples). From this study, we selected a top 39 DEGs from the GEO2R bioinformatics analysis (https://www.ncbi.nlm.nih.gov/geo/geo2r/) based on p value (p < 0.05). The pre-programmed AI and ML algorithm with R programming language was used to generate results for our study (supplementary results: R Script used to generate GEO2R Results)
Determination of gene functions and biological pathways associated with the genes
The 39 DEGs were further analyzed for their functional enrichment to determine which biological functions or pathways they were mostly associated with or enriched in. This functional enrichment analysis was conducted using the ShinyGo bioinformatics tool (http://bioinformatics.sdstate.edu/go/).
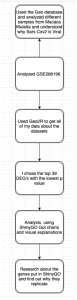
Figure 1: Flowchart. This flowchart summarizes the workflow of the methods used in this research study.
RESULTS
We downloaded the gene expression profile of GSE268196 from the GEO database and used the GEO2R interface connecting the R programming language with the Gene Expression Omnibus (GEO) database to generate our results (Figure 2). We got the graphs by categorizing and analyzing the gene expression profile in the previous study dataset GSE268196. The pre-programmed R Script that generated results in this study is included in the supplementary results: R Script used to generate GEO2R Results.
The GEO2R analysis of the genes expressed differently (DEGs) in the control and infected samples produced several results including a volcano plot and venn diagram as shown in Figure 2 (A, B, C and D). DEGs are genes that show big changes in their activity or gene expression levels under different conditions (Anjum et al., 2016). These conditions can be different tissues, treatments, diseases, stages of development, or environmental factors (Anjum et al., 2016). In Figure 2 the black dots represent the genes that are not expressed differently. Adding on to that, the red dots show genes that are up regulated, and blue represents genes that are down regulated.
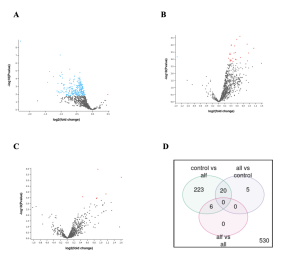
Figure 2: Genes differentially expressed between control samples and samples infected with the virus in Rhesus monkey. Monkeys infected with Sars Cov2 were divided into 3 groups: ALF, ALL, and control (no infection). 2A) ALF vs CONTROL. 2B) ALLvsCONTROL – 2C) ALL vs ALF-ALL and ALF. 2D) Venn diagram shows the total number of differentially expressed genes (DEGs) when comparing the different combinations of infected monkey samples and controls (not infected). A total of 233 DEGs were found in the ALF vs CONTROL sample groups, and 5 in the ALL vs CONTROL sample groups. While there were zero DEGs in the ALF vs ALL sample groups.
In summary, figure 2 shows that some genes were indeed expressed differently between the samples and the controls as shown by the red and blue dots (genes) and the numbers in the venn diagram.
Further Analysis of DEGs to determine Functions or Pathways the DEGs are Enriched in
The number of DEGs obtained in Figure 2 were then narrowed down by identifying the top 39 DEGs using the statistic, P-value (Top 39 DEGs). DEGs are genes that show big changes in their activity levels under different conditions (Anjum et al., 2016). These conditions can be different tissues, treatments, diseases, stages of development, or environmental factors (Anjum et al., 2016). In this study, a gene’s activity was considered important if the P-value was less than 0.05. From these, the top 39 were chosen for further analysis using ShinyGo bioinformatics tool (http://bioinformatics.sdstate.edu/go/).
ShinyGo showed results of what pathways the DEGs were enriched in (Figure 3), the relationship between significantly enriched pathways (Figure 4) and correlation among significant pathways (Figure 5). Results show that the top DEG are mostly associated with or enriched in the pathways, mitogen-activated protein (MAP) kinase, ERK1/2 and Mitogen-activated protein (fold enrichment about 450).

Figure 3-Chart-network enrichment of top DEGs: This fold enrichment chart shows that the top DEG are mostly associated with or enriched in the pathways, mitogen-activated protein (MAP) kinase, ERK1/2 and Mitogen-activated protein (fold enrichment about 450).
Further, these enriched pathways showed connections with each other. Specifically, the pathways mitogen-activated protein (MAP) kinase phosphatase and Mitogen-activated protein Kinase, Transferase, and Protein Kinase were significantly enriched and connected with each other by sharing 20% or more genes (Figure 4).
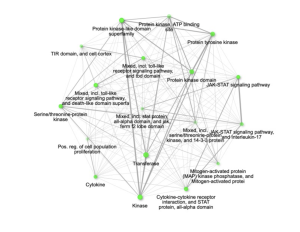
Figure 4: Relationship between enriched pathways. The significantly enriched pathways that are connected and share 20% or more genes are mitogen-activated protein (MAP) kinase phosphatase and Mitogen-activated protein, Kinase, Transferase, and Protein Kinase.Two pathways (nodes) are connected if they share 20% (default) or more genes. Darker nodes are more significantly enriched gene sets. Bigger nodes represent larger gene sets. Thicker edges represent more overlapped genes.
Although ShinyGo was not able to display or specify any genes in the results, further correlation among significant pathways (Figure 5) shown in a Hierarchical clustering tree of clustered pathways indicate that the pathways have many shared genes.

Figure 5-Hierarchical clustering tree: Summary of the correlation among significant pathways listed. Pathways with many shared genes are clustered together. Bigger dots indicate more significant P-values. Clustered together are pathways related to protein kinase, JAK-STAT signaling pathway, and interleukin, Mixed including Toll-lke receptor signaling pathway and Cytokinin-cytokinin receptor interaction, STAT protein, all alpha domain.
Overall, Table 1 summarizes the results from the enrichment pathways of the top 39 DEGs (Figure 3), the relationship between significantly enriched pathways (Figure 4) and correlation among significant pathways (Figure 5).
Table 1. Summary of significantly enriched pathways and their correlation with each other.
Pathways Significantly Enriched | Function and Potential link to SARS-2-Cov-2 |
mitogen-activated protein kinase MAPK | MAPK sends the intracellular signals in the body. Sars Cov2 or Covid19 links to MAPK because when someone is infected with Covid19 the MAPK is activated in their inflammatory system. |
Mitogen-activated protein | Sends intracellular signals throughout the body. Sends the signals when someone has covid. |
ERK1/2 | Regulates stimulated cellular processes. When the MAPK is activated when someone is infected with SARS Cov2, the ERK1/2 is also triggered in the process. |
Interleukin17 | Interleukin17 activates and mobilizes the cell. When there is a higher Interleukin level the chance of catching Covid19 is higher. |
JAK STAT signaling pathway | Binds DNA and allows the transcription of genes. This is linked to Covid 19 because it amplifies the pathological effects of Covid 19. |
DISCUSSION
The purpose of the research was to find out why viruses spread or replicate so quickly. The scientific question being investigated was why the SARS-Cov-2 virus replicated so fast, and how they do so in relation to the genes that are expressed during infection. SARS-CoV-2 replicates virally through the lungs and leads to different clinical outcomes. This can include people who are asymptomatic, to people who are near death (W. Joost Wiersinga p et al 2020). Inflammation of the lungs happens during severe infection (Duan et al, 2024). And as we know, COVID-19 was one of the biggest pandemics that humans ever faced.
A previous study showed results that suggested that early-stage COVID-19 should be treated by trying to stop the virus from replicating, while later-stage therapy should focus on reducing inflammation (Duan et al, 2024). This study provided new information into why SARS-CoV-2 initially causes mild symptoms but can become potentially deadly for some patients about a week after infection. The researchers demonstrated that different stages of illness are linked to the virus behaving differently in two distinct groups of cells (Duan et al, 2024 ). The team discovered that when SARS-CoV-2 infects lung lining cells initially, two viral proteins operate within these cells. One protein activates the immune system, while another paradoxically blocks this activation, leading to minimal inflammation (Duan et al, 2024). Additionally, the virus can use a different pathway to enter immune cells. This pathway limits viral replication and prevents the production of the immune signal-blocking protein. As a result, the first protein can trigger excessive inflammation associated with severe symptoms (Duan et al, 2024).
Therefore, to find out what genes are differentially expressed and linked to the rapid replication of SARS-CoV-2, we went to various bioinformatics databases and tools to collect data and conducted the bioinformatics research (Figure 1). Our goal was to go a step further and potentially identify genes that are differentially expressed and linked to the rapid replication spreading of SARS-CoV-2.
The bioinformatics database used in this study was GSE268196 where researchers originally collected preserved lung cells from rhesus macaques (monkeys) infected with SARS-CoV-2 on days 0, 3, 7, 14, and 28-35 after infection. They had 71 samples in total, divided into three groups: aIFNg (22 samples), aIL10 (24 samples), and control (24 samples) (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE268196). These scientists’ results suggested that SARS-CoV-2 might spread more easily in lungs with less immune activity and that recent lung inflammation can affect the severity of COVID-19 across different people.
However in our bioinformatics study, we did not see any of our top DEGs enriched in immunity. Instead, our results showed that the top DEGs were significantly enriched in the biological pathways related to inflammation; mitogen-activated protein (MAP) kinase (MAPK) phosphatase and Mitogen-activated protein. Kinase, Transferase(no previous study that links to Sars Cov2), and Protein Kinase (Figures 3, 4 and 5). This is similar to a previous study by who found that MAPK-related biomarker levels were higher in participants who tested positive for SARS-CoV-2 (89 people) compared to those who tested negative (29 people) (Cusato et al, 2023). The researchers therefore suggested that MAPK-related biomarkers might influence COVID-19 symptoms and could help identify people with COVID-19 who are at risk for inflammation complications.
In addition, the presence of these MAPK enriched inflammation-related pathways in our results is supported by a previous study that showed that SARS-CoV-2 can infect key immune cells, triggering strong inflammatory signals (Junqueira et al, 2022). The researchers examined fresh blood samples from COVID-19 patients at Massachusetts General Hospital’s emergency department. They compared these samples with those from healthy people and patients with other respiratory conditions. They also studied lung tissue from autopsies of people who died from COVID-19 (Junqueira et al, 2022). The team discovered that SARS-CoV-2 can infect two types of immune cells that act as early responders to infection: monocytes in the blood and macrophages in the lungs. When these cells become infected, they die a fiery death called pyroptosis. As they die, they release a burst of powerful inflammatory alarm signals (Junqueira et al, 2022).
Therefore, the results in our study can be used to further analyze the potential use of these inflammation MAPK-related proteins and pathways as anti-inflammatory medication for COVID-19 patients.
Limitations
Because our research used data from experiments done by other researchers, one limitation is that the enriched pathways we found in this study will need to be studied more in the laboratory or in a clinical setting before anti-inflammatory medication can be potentially developed in future studies.
Conclusion
The goal of the research was to identify genes that are expressed differently and how they can help explain why SARS-CoV-2 viruses replicate so quickly. Before the research was started, the hypothesis was that the SARS-CoV-2 virus replicates by taking over the host’s gene expression machinery and upregulating genes related to replication. However, our study shows that instead, the genes that were differentially expressed in hosts infected with SARS-CoV-2, were more associated with proteins and pathways related to inflammation, including the mitogen-activated protein kinase (MAPK) pathway. When a person is infected with the SARS COV2 virus, their inflammatory system is impacted. When this happens the MAPK sends a signal to the rest of the body. Since COVID-19 often causes uncontrolled inflammatory responses through pathways like MAPK, these proteins and pathways shown in our study could be targeted in future studies to provide customized anti-inflammatory treatments for COVID-19 patients, in addition to the regular early use of antiviral drugs.
Supplementary Results
References
- Anjum, A., Jaggi, S., Varghese, E., Lall, S., Bhowmik, A., & Rai, A. (2016, April 1). Identification of differentially expressed genes in RNA-seq data of Arabidopsis thaliana: A compound distribution approach. Journal of computational biology : a journal of computational molecular cell biology.
- Barrett, T., Suzek, T. O., Troup, D. B., Wilhite, S. E., Ngau, W. C., Ledoux, P., … & Edgar, R. (2005). NCBI GEO: mining millions of expression profiles—database and tools. Nucleic acids research, 33(suppl_1), D562-D566.
- Chenchula S., Karunakaran P., Sharma S., Chavan M. Current evidence on efficacy of COVID-19 booster dose vaccination against the Omicron variant: A systematic review. J Med Virol. 2022 Jul;94(7):2969-2976.
- Cusato J, Manca A, Palermiti A, Mula J, Costanzo M, Antonucci M, Trunfio M, Corcione S, Chiara F, De Vivo ED, Ianniello A, Ferrara M, Di Perri G, De Rosa FG, D’Avolio A, Calcagno A. COVID-19: A Possible Contribution of the MAPK Pathway. Biomedicines. 2023 May 16;11(5):1459. doi: 10.3390/biomedicines11051459. PMID: 37239131; PMCID: PMC10216575.
- Duan, T., Xing, C., Chu, J. et al. ACE2-dependent and -independent SARS-CoV-2 entries dictate viral replication and inflammatory response during infection. Nat Cell Biol 26, 628–644 (2024). https://doi.org/10.1038/s41556-024-01388-w
- Ge, S. X., Jung, D., & Yao, R. (2020). ShinyGO: a graphical gene-set enrichment tool for animals and plants. Bioinformatics, 36(8), 2628-2629. (http://bioinformatics.sdstate.edu/go/)
- Geall, A. J., Mandl, C. W., Ulmer, J. B. RNA: The new revolution in nucleic acid vaccines. Semin Immunol. 2013;25(2):152–159. doi: 10.1016/j.smim.2013.05.001.
- Hu, B., Guo, H., Zhou, P. et al. Characteristics of SARS-CoV-2 and COVID-19. Nat Rev Microbiol 19, 141–154 (2021). https://doi.org/10.1038/s41579-020-00459-7
- Junqueira, C., Crespo, Â., Ranjbar, S. et al. FcγR-mediated SARS-CoV-2 infection of monocytes activates inflammation. Nature 606, 576–584 (2022). https://doi.org/10.1038/s41586-022-04702-4
- Kim, T. H., Choi, S. J., Lee, Y. H., Song, G. G., & Ji, J. D. (2014). Gene expression profile predicting the response to anti-TNF treatment in patients with rheumatoid arthritis; analysis of GEO datasets. Joint Bone Spine, 81(4), 325-330. (GEO2R)
- Lundstrom, K. Replicon RNA viral vectors as vaccines. Vaccines. 2016;4:39. doi: 10.3390/vaccines4040039.
- Philip V Kovski, Annika Kratzil, Silvio Steiner, Hanspeter Stalder, Volker Thiel. Corona Virus and Replication: Implications for SARS-CoV-2
- Rawaa S. Al-Kayali, Mohomed F. Kashkash, Azzam H. Alhussein Alhajji, Abdullah Khouri. Activation of tuberculosis in recovered Covid-19 patients: a case report, 2023
- Raman R., Patel K. J., Ranjan K. COVID-19: Unmasking Emerging SARS-CoV-2 Variants, Vaccines and Therapeutic Strategies. Biomolecules. 2021 Jul 06;11(7)
- Rosa, S. S., Prazeres, D. M., Azevedo, A. M., & Marques, M. P. (2021). mRNA vaccines manufacturing: Challenges and bottlenecks. Vaccine, 39(16), 2190-2200.
- Rosa SS, Prazeres DMF, Azevedo AM, Marques MPC. mRNA vaccines manufacturing: Challenges and bottlenecks. Vaccine. 2021 Apr 15;39(16):2190-2200. doi: 10.1016/j.vaccine.2021.03.038. Epub 2021 Mar 24. PMID: 33771389; PMCID: PMC7987532.
- Ross, J. mRNA stability in mammalian cells. Microbiol Mol Biol Rev. 1995;59:423–450.
- Schlake, T., Thess, A., Fotin-Mleczek, M., Kallen, K.-J. Developing mRNA-vaccine technologies. RNA Biol. 2012;9(11):1319–1330. doi: 10.4161/rna.22269
- Schoch, C. L., Ciufo, S., Domrachev, M., Hotton, C. L., Kannan, S., Khovanskaya, R., … & Karsch-Mizrachi, I. (2020). NCBI Taxonomy: a comprehensive update on curation, resources and tools. Database, 2020, baaa062.
- Sharma A., Ahmad Farouk I., Lal SK. COVID-19: A Review on the Novel Coronavirus Disease Evolution, Transmission, Detection, Control and Prevention. Viruses. 2021 Jan 29;13(2)
- Vogel A. B., Lambert L., Kinnear E., Busse D., Erbar S., Reuter K. C., et al. Self-Amplifying RNA Vaccines Give Equivalent Protection against Influenza to mRNA Vaccines but at Much Lower Doses. Mol Ther. 2018;26(2):446–455. doi: 10.1016/j.ymthe.2017.11.017
- Walensky RP, Walke HT, Fauci AS. SARS-CoV-2 Variants of Concern in the United States-Challenges and Opportunities. JAMA. 2021 Mar 16;325(11):1037-1038.
- Wiersinga W. J., Rhodes A., Cheng A. C., Peacock S. J., Prescott H. C. Pathophysiology, Transmission, Diagnosis, and Treatment of Coronavirus Disease 2019 (COVID-19): A Review. JAMA. 2020;324(8):782–793. doi:10.1001/jama.2020.12839
- Zhang Y, Bharathi V, Dokoshi T, de Anda J, Ursery LT, Kulkarni NN, Nakamura Y, Chen J, Luo EWC, Wang L, Xu H, Coady A, Zurich R, Lee MW, Matsui T, Lee H, Chan LC, Schepmoes AA, Lipton MS, Zhao R, Adkins JN, Clair GC, Thurlow LR, Schisler JC, Wolfgang MC, Hagan RS, Yeaman MR, Weiss TM, Chen X, Li MMH, Nizet V, Antoniak S, Mackman N, Gallo RL, Wong GCL. Viral afterlife: SARS-CoV-2 as a reservoir of immunomimetic peptides that reassemble into proinflammatory supramolecular complexes. Proc Natl Acad Sci U S A. 2024 Feb 6;121(6):e2300644120. doi: 10.1073/pnas.2300644120. Epub 2024 Feb 2. PMID: 38306481; PMCID: PMC10861912.

