Identification of Key Genes and Pathways in Clade I Monkeypox Virus to Find Possible Treatments using Bioinformatics

ABSTRACT
Background
The virus being studied is Clade I Monkeypox. It can cause fever, rashes, lymphadenopathy, and other skin related symptoms if not treated. It spreads through direct contact rapidly which led to major global outbreaks in 2022 and 2023.The research on how the virus affects gene activity in host cells in limited, created a gap in knowledge when developing treatments. This study aims to identify differentially expressed genes in keratinocytes exposed to Clade I Monkeypox and examine their functions and pathways.
Methods
We used the NCBI GEO dataset GSE219036, which compares gene expression in keratinocytes and colon organoids infected with Clade I and Clade II Monkeypox. The result of the original study was that Clade II affected hypoxia related genes. Using GEO2R, we identified differentially expressed genes in Clade I infected keratinocytes and selected 30 samples using p values and log2 fold change. These genes were analyzed with Shiny Go for KEGG and GO enrichment.
Results
From 15,407 genes, we found the 15 most upregulated and downregulated differentially expressed genes. KEGG analysis showed enrichment in three pathways, with systemic lupus erythematosus being the most enriched. The genes H2A and H4 were found as significant in this pathway. GO analysis revealed enrichment in four pathways but now specific genes were found.
Conclusion
In keratinocytes infected with Clade I Monkeypox, H2A and H4 were differentially expressed and are linked to autoimmune responses. The GO results suggest the virus disrupts the cellular structure. These results may contribute to developing new treatments.
INTRODUCTION
Monkeypox is a viral infection whose symptoms are a fever, rash, and lesions (1). It is spread through close contact and was initially restricted to Central and West Africa. In comparison to Clade II strains of Monkeypox, Clade I has increased disease severity and higher mortality rates (2).
Even though monkeypox has been around since 1958, there is lack of research about how the more severe Clade I strain affects human skills at the gene level. The reason for this is that the Clade I strain, until recently, has remained endemic in Africa. The more recent Clade IIb strain, which was responsible for the 2022 outbreak, has been the focus of recent research . Even though efforts have been made to further research Clade I Monkeypox, it still remains understudied compared to the Clade II strains of Monkeypox (3). This project addresses this knowledge gap by analyzing data on gene expression in keratinocytes that were infected with the Clade I strain.
The scientific question being investigated is; how is the gene expression affected in human skin cells, or keratinocytes, that are infected with the Clade I monkeypox virus and which biological pathways are most disrupted compared to uninfected skin cells.
What is known so far is that the Clade I Monkeypox strain has caused fatality rates as high as 10.6% percent, especially being prominent in areas with limited healthcare access. Recent cases in the U.S. show that it can spread globally. It can cause strong immune reactions and tissue damage (1). In addition, the majority of bioinformatics studies have been conducted on Clade IIb, with tools like GEO2R being used to analyze differential gene expression and immune response. Transcriptomics analysis of Clade I is limited, however. There is a paucity of data about how the Clade I Monkeypox strain affects a host’s genes or cellular pathways. This makes it difficult to predict the severity of the disease in a person and makes it hard to identify molecular targets (1,4).
The goal of this research is to examine gene expression changes due to Clade I infection in keratinocytes through publicly accessible data and bioinformatics platforms, with the aim of identifying target genes and pathways. I hypothesize that the Clade I infection will noticeably change the gene expression in genes involved in immune signaling, inflammation, and stress responses for skin cells.
By finding the gene-level impacts of Clade I, this study can improve our knowledge of the Clade I strain of Monkeypox, support research towards finding a vaccine for it, and help prepare for future outbreaks.
METHODS
Data Collection and Analysis of GEO2R Data
To start my investigation, I went to the National Center for Biotechnology Information and looked for GEO Datasets about Monkeypox. GEO2R is an online tool that is offered through the National Center for Biotechnology Information. It helps analyze gene expression across various samples (5). Eventually, we selected the dataset GSE219036 which is called “Virological characterization of the 2022 outbreak-causing monkeypox virus using human keratinocytes and colon organoids”. This data set contains RNA sequence data for infected and uninfected human keratinocyte samples. Figure 1 summarizes the methods and bioinformatics tools used in this study.
Figure 1: Research Methodology: The steps and bioinformatics used in this study.
Identification of the Top Differentially Expressed Genes
To identify the top 30 most significant differentially expressed genes, statistical analysis was applied. This process used both p value and the Fold change value. First, all samples with p values over 0.05 were eliminated. Next, 15 samples with the highest Fold change value and 15 samples with the lowest Fold change value were selected. This process helped find the 30 most significant differentially expressed genes.
Functional and Enrichment Analysis Using SRPlot, KEGG, and GO Bioinformatics Tools
Then Shiny GO, KEGG, and GO bioinformatics tools and databases were utilized to analyze the functions of these top genes(6). These tools helped uncover the potential roles of the genes in specific molecular pathways, biological processes, and disease processes. First, the gene IDs from the top 30 differentially expressed genes were entered into Shiny GO for enrichment analysis. Through Shiny GO, I used KEGG to identify 3 specific pathways. The molecular pathways found were Systemic Lupus Erythematosus, Alcoholism, and Neutrophil Extracellular Trap Formation. Through the Systemic Lupus Erythematosus pathway, the most relevant out of the three, I was able to identify the H2A and H4 genes as being the most significant. Using the GO biological process, I was able to identify 4 specific pathways. The molecular pathways found were intermediate filament organization, keratinization, intermediate filament cytoskeleton organization, and intermediate filament based process. However, no specific genes were able to be identified.
RESULTS
Identification of Differentially Expressed Genes
To identify differentially expressed genes I used GEO2R. From my first two results, 10812 genes out of the15407 genes total expressed differently between the two groups of the control keratinocytes and keratinocytes inflected with Clade I Monkeypox.
From my volcano plot (Figure 2a), the red dots represent upregulated genes, or genes with higher expression levels, in the infected keratinocytes compared to the control skin cells while the blue dots represent down regulated genes, or genes with lower expression levels, in the infected cells as opposed to the normal keratinocytes.
From the venn diagram (Figure 2b), in total the study produced 15,407 genes. Out of these genes, there were 10,812 differentially expressed genes that overlapped between the keratinocytes that were infected with Clade I Monkeypox and the control cells. This large overlap shows that the keratinocytes have a strong biological response against the virus.
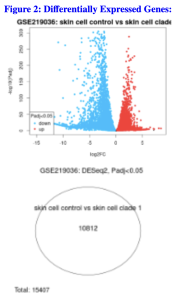
Figure 2. Identification of Differentially Expressed Genes. a) The volcano plot shows the distribution of the differentially expressed genes. The genes in red are the upregulated genes and the genes in blue are the down regulated genes. b) The venn diagram shows that 10,812 genes out of the 15,407 genes are shared between the control and infected keratinocytes.
Identification of 30 Statistically Significant Differentially Expressed Genes (DEGs)
To narrow down my genes, I used both the p value and the fold change value. Using these, I narrowed my list to 30 differentially expressed genes. I chose 15 upregulated genes and 15 down regulated genes. (Top 30 DEGs)
Potential Functions and Enrichment of the Identified Genes and/or pathways
I used ShinyGo to determine the potential functions of the genes. From the KEGG results, I identified Systemic Lupus Erythematosus, Alcoholism, and Neutrophil Extracellular Trap Formation. Out of these three, I found System Lupus Erythematosus as the most significant pathway as it had the highest Fold Enrichment value (Figure 3a). From this pathway, the genes that stood out were the H2A gene and the H4 gene (Figure 3b).
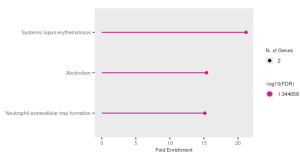
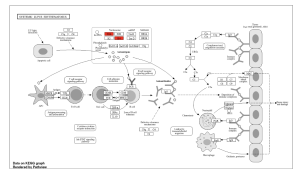
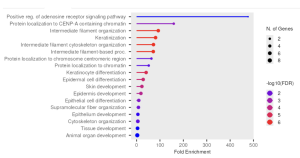
Figure 3. Functional and Enrichment Analysis. a) This figure shows the three significant pathways found through KEGG with Systemic Lupus Erythematosus being the most significant out of the three.3b. This figure shows the Systemic Lupus Erythematosus pathway in depth, with the genes H2A and H4 (highlighted in red) being the most significant. c) This figure shows the GO significant pathways in red.
From the GO results, I identified intermediate filament organization, keratinization, intermediate filament cytoskeleton organization, and intermediate filament based process (Figure 3c). However, no specific genes stood out. Table 1 below summarizes the key genes and pathways identified in this study.
Table 1: Summary of Key Pathways and Genes Identified in this Study
Functional and Enrichment Analysis | Key Pathways | Genes |
KEGG | Systemic Lupus Erythematosus, Alcoholism, and Neutrophil Extracellular Trap Formation | H2A and H4 |
GO | intermediate filament organization, keratinization, intermediate filament cytoskeleton organization, and intermediate filament based process | N/A |
DISCUSSION
Summary of Findings
The goal of this research study was to identify differentially expressed genes in keratinocytes associated with Clade I Monkeypox and analyze their biological functions using bioinformatics tools. From the GEO dataset GSE219036, a total of 15,407 genes were analyzed (Figure 2b). Using a venn diagram, it was seen that 10,812 of these genes were common across both the control group and the infected group (Figure 2b). After using statistical analysis such as p value and fold enrichment to eliminate insignificant genes, the list was narrowed down to the top 30 differentially expressed genes. With ShinyGO, a number of significantly enriched pathways were obtained from the KEGG and GO Biological Processes pathways. The top enriched KEGG pathways were systemic lupus erythematosus, alcoholism, and neutrophil extracellular trap formation (Figure 3a). Specifically in the systemic lupus erythematosus pathways, the most significant pathway out of the three, the genes H2A and H4 stood out (Figure 3b). The top enriched GO pathways were intermediate filament organization, keratinization, intermediate filament cytoskeleton organization, and intermediate filament based process (Figure 3c).
Interpretation of Results
GEO2R analysis results indicate significant gene expression changes in Clade I Monkeypox-infected keratinocytes. The top 30 differentially expressed genes indicate that they could be of significance in host response to monkeypox infection. The KEGG analysis indicated that these genes were primarily associated with pathways in systemic lupus erythematosus, alcoholism, and neutrophil extracellular trap formation (Figure 3a). Out of these three, systemic lupus erythematosus stood out since it had the highest fold enrichment (Figure 3a). This pathway shows that Monkeypox interferes with the host’s autoimmune system (7). The occurrence of H2A and H4 within the systemic lupus erythematosus pathway is indicative of changes in how DNA is organized during infection. The GO biological process results also supported such findings by identifying processes such as keratinization and intermediate filament organization, which are highly relevant in keratinocytes’ cellular structure. This indicates the virus might interfere with the cytoskeleton of skin cells as a part of its attack mechanism.
Comparison with Other Studies
Other studies have also had similar findings. A study that wanted to examine if there was a link between Monkeypox and Lupus Nephritis, a form of Systemic Lupus Erythematosus found in kidneys, by using bioinformatics. They found that both diseases showed similar gene expression, meaning that Monkeypox could influence or worsen Lupus Nephritis (8). This is in line with the KEGG enrichment results. Other studies have also highlighted how Monkeypox affects keratinization and disrupts cellular structural integrity (9,10). This supports the GO enrichment results, which show gene expression in these two areas.
Implications
This study provides insight on how gene expression in skin cells change during their immune response to Monkeypox. Practical applications of these findings may include identifying biomarkers for early detection of Monkeypox and helping predict the severity of the virus. By specifically identifying the genes H2A and H4, epigenetic therapies, therapies that involve turning on and off certain genes, and targeted drugs that regulate gene activity become a possibility. These findings may contribute and expedite the process for the development of a vaccine or antiviral treatments against Monkeypox.
Limitations
In our case, since we performed secondary research and used bioinformatics datasets from experiments conducted by other researchers, one limitation is that the identified genes and pathways require further research in a laboratory or clinical environment before developing a vaccine.
Future Direction
The identified genes can be tested in the laboratory by scientists in the laboratory or clinical trials to determine if they play a direct role in how Monkeypox Clade I affects keratinocytes. Through further study, researchers can deduce whether these genes can serve as accurate biomarkers for the virus and their relevance in the development of treatments. Further experiments can also aim to see how these genes are expressed over time of an infection or if they are active in different strains of Monkeypox.
References
- Gu, C., Huang, Z., Sun, Y., Shi, S., Li, X., Li, N., Liu, Y., Guo, Z., Jin, N., Zhao, Z., Li, X., & Wang, H. (2024). Characterization of human immortalized keratinocyte cells infected by monkeypox virus. Viruses, 16(8), 1206. https://pmc.ncbi.nlm.nih.gov/articles/PMC11359611/
- Levy V, Branzuela A, Hsieh K, et al. First Clade Ib Monkeypox Virus Infection Reported in the Americas — California, November 2024. MMWR Morb Mortal Wkly Rep 2025;74:44–49. http://dx.doi.org/10.15585/mmwr.mm7404a1
- Alakunle E, Kolawole D, Diaz-Cánova D, Alele F, Adegboye O, Moens U and Okeke MI (2024) A comprehensive review of monkeypox virus and mpox characteristics. Front. Cell. Infect. Microbiol. 14:1360586. doi: 10.3389/fcimb.2024.1360586
- Patel VM, Patel SV. Epidemiological Review on Monkeypox. Cureus. 2023 Feb 5;15(2):e34653. doi: 10.7759/cureus.34653. PMID: 36895541; PMCID: PMC9991112.
- Emily Clough, Tanya Barrett, Stephen E Wilhite, Pierre Ledoux, Carlos Evangelista, Irene F Kim, Maxim Tomashevsky, Kimberly A Marshall, Katherine H Phillippy, Patti M Sherman, Hyeseung Lee, Naigong Zhang, Nadezhda Serova, Lukas Wagner, Vadim Zalunin, Andrey Kochergin, Alexandra Soboleva, NCBI GEO: archive for gene expression and epigenomics data sets: 23-year update, Nucleic Acids Research, Volume 52, Issue D1, 5 January 2024, Pages D138–D144, https://doi.org/10.1093/nar/gkad965
- Rodriguez A, Getino A. BioFunctional: A Comprehensive App for Interpreting and Visualizing Functional Analysis of KEGG Pathways and Gene Ontologies [Internet]. biorxiv. biorxiv; 2024 [cited 2025 Aug 6]. https://www.biorxiv.org/content/10.1101/2024.10.08.616405v1.abstract
- Nakano M, Ota M, Yusuke Takeshima, Iwasaki Y, Hatano H, Yasuo Nagafuchi, et al. Distinct transcriptome architectures underlying lupus establishment and exacerbation. Cell [Internet]. 2022 Aug 22 [cited 2024 Nov 18];185(18):3375-3389.e21.
- Wang Y, Li Q. Integrative bioinformatics analysis reveals STAT1, ORC2, and GTF2B as critical biomarkers in lupus nephritis with Monkeypox virus infection. Scientific Reports [Internet]. 2025 Apr 19 [cited 2025 Aug 7];15(1). Available from: https://www.nature.com/articles/s41598-025-97791-w
- Watanabe Y, Kimura I, Hashimoto R, Sakamoto A, Yasuhara N, Yamamoto T, et al. Virological characterization of the 2022 outbreak‐causing monkeypox virus using human keratinocytes and colon organoids. 2023 Jun 1;95(6).
- Witt ASA, Trindade G de S, Souza FG de, Serafim MSM, da Costa AVB, Silva MVF, et al. Ultrastructural analysis of monkeypox virus replication in Vero cells. Journal of Medical Virology [Internet]. 2023 Feb 1;95(2):e28536. Available from: https://pubmed.ncbi.nlm.nih.gov/36708101/

